Create a beautifully simple personal or academic website in under 10 minutes.
Selected Publications
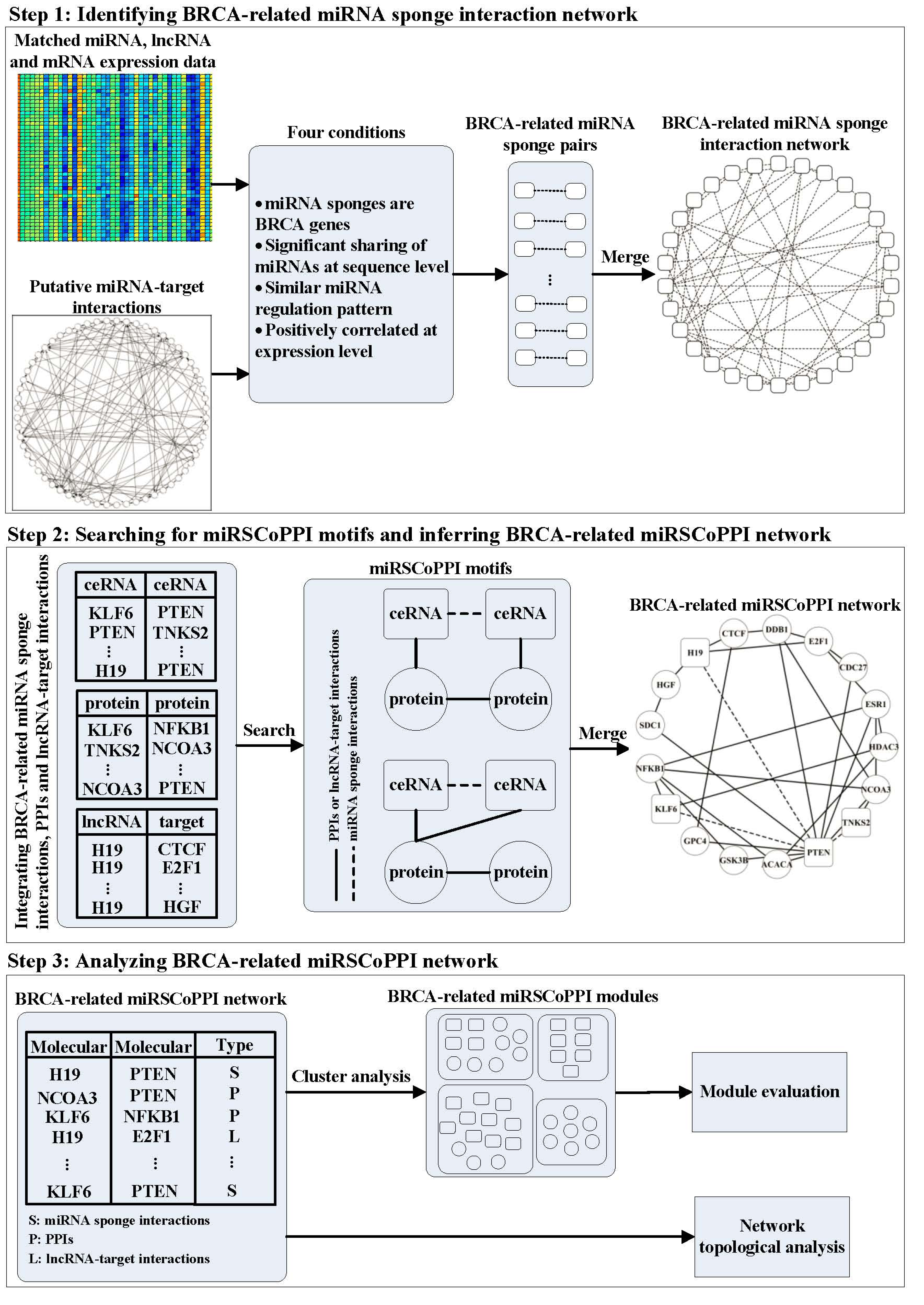
Inferring miRNA sponge co-regulation of protein-protein interactions in human breast cancer
Background Recent studies have shown that the crosstalk between microRNA (miRNA) sponges plays an important role in human cancers. However, the co-regulation roles of miRNA sponges in protein-protein interactions (PPIs) are still unknown. Results In this study, we propose a multi-step method called miRSCoPPI to infer miRNA sponge co-regulation of PPIs. We focus on investigating breast cancer (BRCA) related miRNA sponge co-regulation, by integrating heterogeneous data, including miRNA, long non-coding RNA (lncRNA) and messenger RNA (mRNA) expression data, experimentally validated miRNA-target interactions, PPIs and lncRNA-target interactions, and the list of breast cancer genes. We find that the inferred BRCA-related miRSCoPPI network is highly connected and scale free. The top 10% hub genes in the BRCA-related miRSCoPPI network have potential biological implications in breast cancer. By utilizing a graph clustering method, we discover 17 BRCA-related miRSCoPPI modules. Through pathway enrichment analysis of the modules, we find that several modules are significantly enriched in pathways associated with breast cancer. Moreover, 10 modules have good performance in classifying breast tumor and normal samples, and can act as module signatures for prognostication. By using putative computationally predicted miRNA-target interactions, we have consistent results with those obtained using experimentally validated miRNA-target interactions, indicating that miRSCoPPI is robust in inferring miRNA sponge co-regulation of PPIs in human breast cancer. Conclusions Taken together, the results demonstrate that miRSCoPPI is a promising tool for inferring BRCA-related miRNA sponge co-regulation of PPIs and it can help with the understanding of the co-regulation roles of miRNA sponges on the PPIs.
BMC Bioinformatics
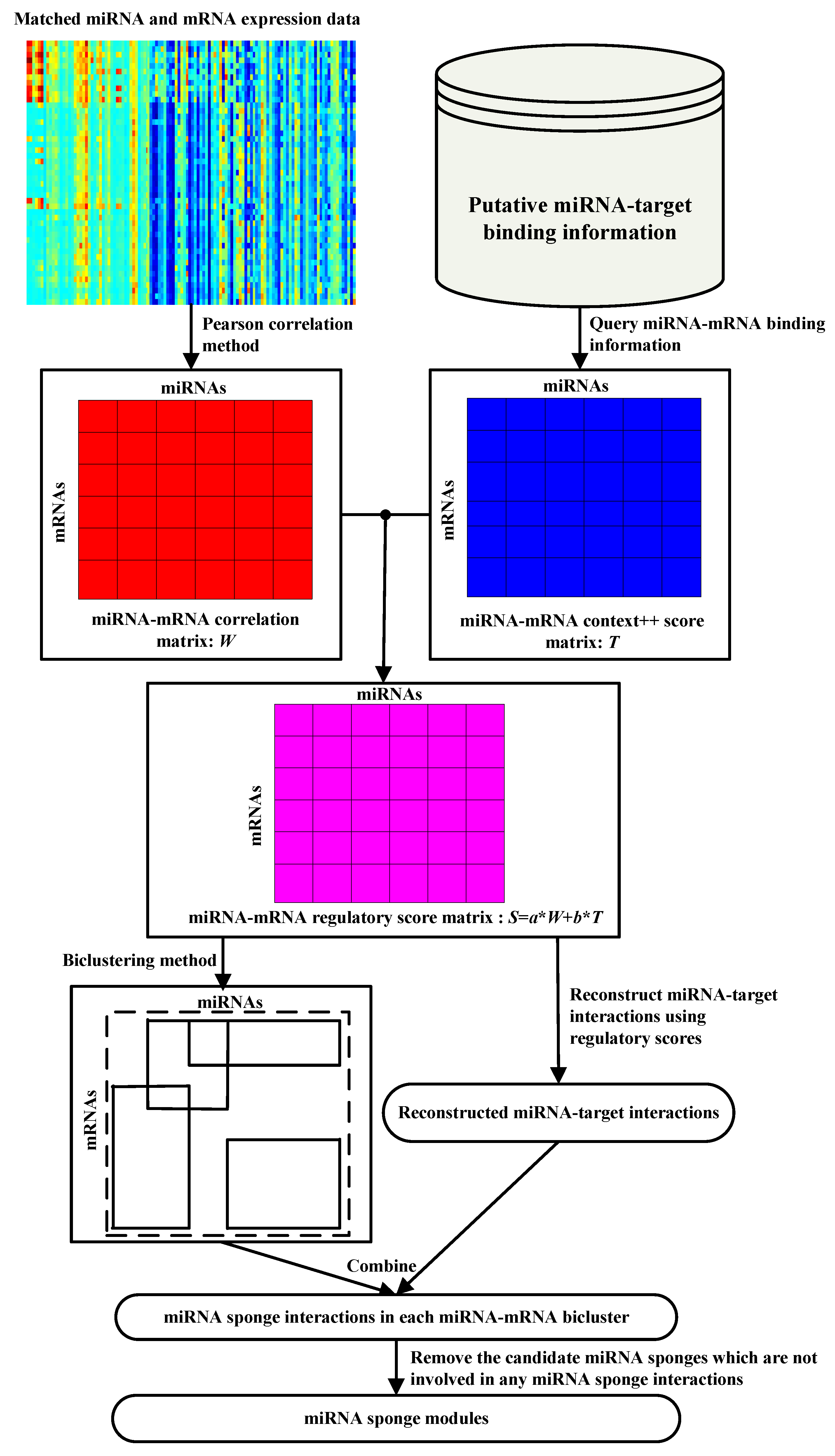
Identifying miRNA sponge modules using biclustering and regulatory scores
Background MicroRNA (miRNA) sponges with multiple tandem miRNA binding sequences can sequester miRNAs from their endogenous target mRNAs. Therefore, miRNA sponge acting as a decoy is extremely important for long-term loss-of-function studies both in vivo and in silico. Recently, a growing number of in silico methods have been used as an effective technique to generate hypotheses for in vivo methods for studying the biological functions and regulatory mechanisms of miRNA sponges. However, most existing in silico methods only focus on studying miRNA sponge interactions or networks in cancer, the module-level properties of miRNA sponges in cancer is still largely unknown. ResultsWe propose a novel in silico method, called miRSM (miRNA Sponge Module) to infer miRNA sponge modules in breast cancer. We apply miRSM to the breast invasive carcinoma (BRCA) dataset provided by The Cancer Genome Altas (TCGA), and make functional validation of the computational results. We discover that most miRNA sponge interactions are module-conserved across two modules, and a minority of miRNA sponge interactions are module-specific, existing only in a single module. Through functional annotation and differential expression analysis, we also find that the modules discovered using miRSM are functional miRNA sponge modules associated with BRCA. Moreover, the module-specific miRNA sponge interactions among miRNA sponge modules may be involved in the progression and development of BRCA. Our experimental results show that miRSM is comparable to the benchmark methods in recovering experimentally confirmed miRNA sponge interactions, and miRSM outperforms the benchmark methods in identifying interactions that are related to breast cancer. Conclusions Altogether, the functional validation results demonstrate that miRSM is a promising method to identify miRNA sponge modules and interactions, and may provide new insights for understanding the roles of miRNA sponges in cancer progression and development.
BMC Bioinformatics
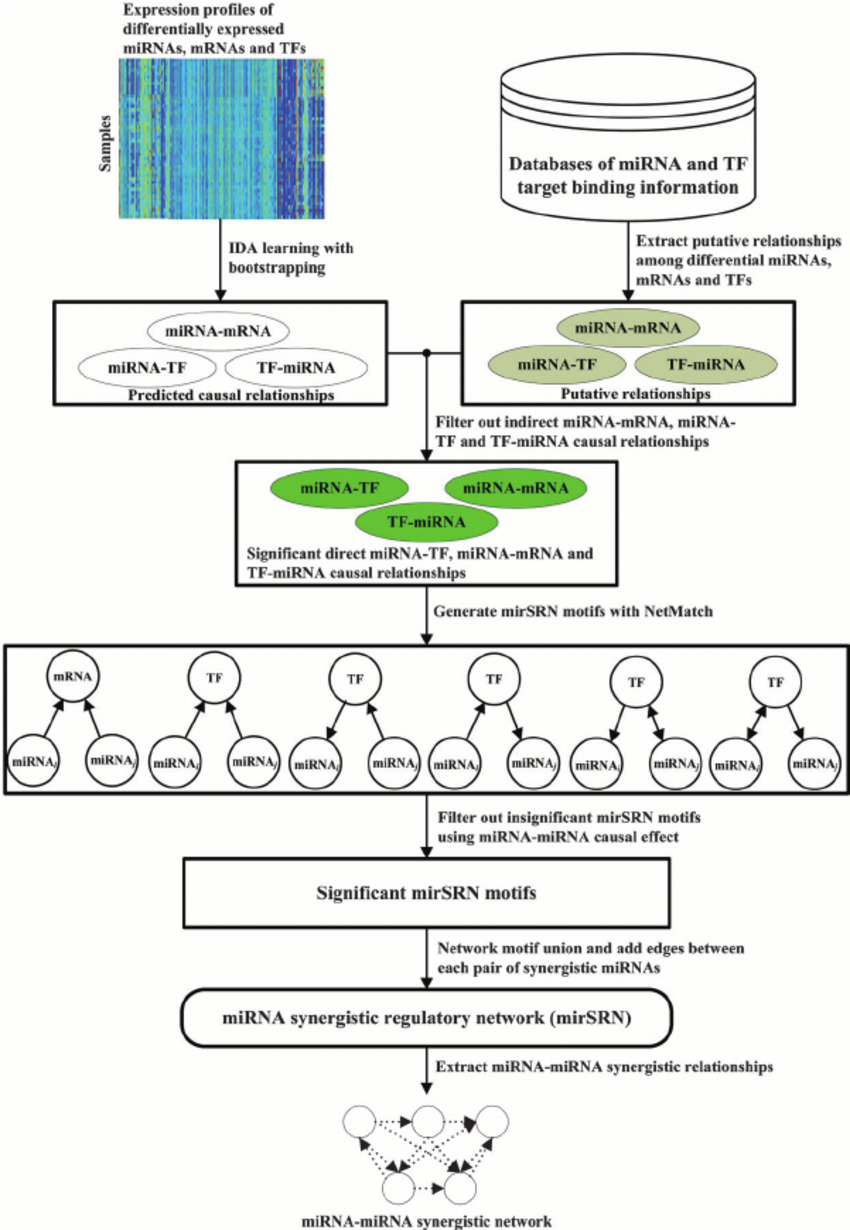
Identifying miRNA synergistic regulatory networks in heterogeneous human data via network motifs
Understanding the synergism of multiple microRNAs (miRNAs) in gene regulation can provide important insights into the mechanisms of complex human diseases caused by miRNA regulation. Therefore, it is important to identify miRNA synergism and study miRNA characteristics in miRNA synergistic regulatory networks. A number of methods have been proposed to identify miRNA synergism. However, most of the methods only use downstream target genes of miRNAs to infer miRNA synergism when miRNAs can also be regulated by upstream transcription factors (TFs) at the transcriptional level. Additionally, most methods are based on statistical associations identified from data without considering the causal nature of gene regulation. In this paper, we present a causality based framework, called mirSRN (miRNA synergistic regulatory network), to infer miRNA synergism in human molecular systems by considering both downstream miRNA targets and upstream TF regulation. We apply the proposed framework to two real world datasets and discover that almost all the top 10 miRNAs with the largest node degree in the mirSRNs are associated with different human diseases, including cancer, and that the mirSRNs are approximately scale-free and small-world networks. We also find that most miRNAs in the networks are frequently synergistic with other miRNAs, and miRNAs related to the same disease are likely to be synergistic and in a cluster linked to a biological function. Synergistic miRNA pairs show higher co-expression level, and may have potential functional relationships indicating collaboration between the miRNAs. Functional validation of the identified synergistic miRNAs demonstrates that these miRNAs cause different kinds of diseases. These results deepen our understanding of the biological meaning of miRNA synergism.
Molecular BioSystems
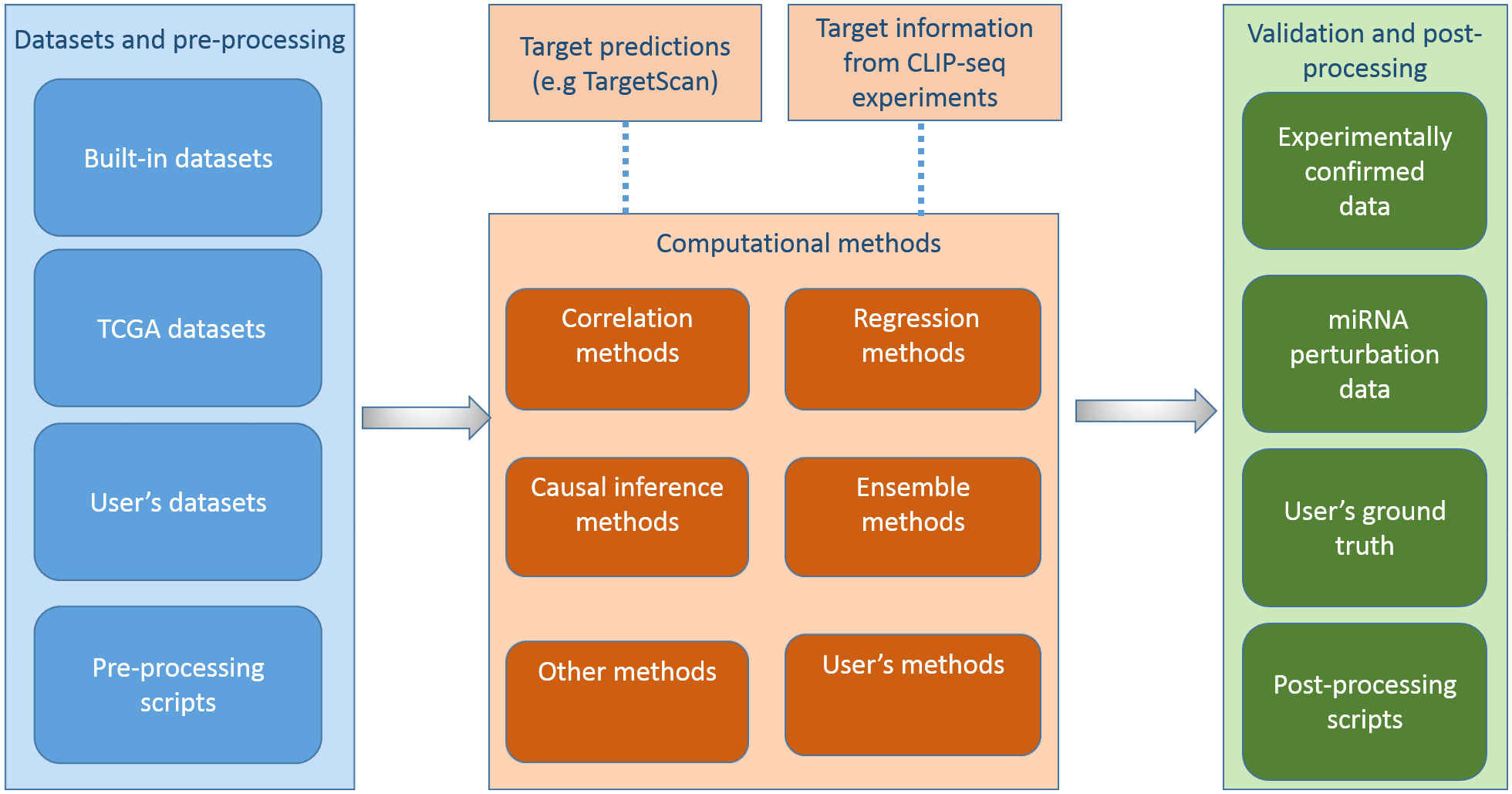
miRLAB: An R Based Dry Lab for Exploring miRNA-mRNA Regulatory Relationships
microRNAs (miRNAs) are important gene regulators at post-transcriptional level, and inferring miRNA-mRNA regulatory relationships is a crucial problem. Consequently, several computational methods of predicting miRNA targets have been proposed using expression data with or without sequence based miRNA target information. A typical procedure for applying and evaluating such a method is i) collecting matched miRNA and mRNA expression profiles in a specific condition, e.g. a cancer dataset from The Cancer Genome Atlas (TCGA), ii) applying the new computational method to the selected dataset, iii) validating the predictions against knowledge from literature and third-party databases, and comparing the performance of the method with some existing methods. This procedure is time consuming given the time elapsed when collecting and processing data, repeating the work from existing methods, searching for knowledge from literature and third-party databases to validate the results, and comparing the results from different methods. The time consuming procedure prevents researchers from quickly testing new computational models, analysing new datasets, and selecting suitable methods for assisting with the experiment design. Here, we present an R package, miRLAB, for automating the procedure of inferring and validating miRNA-mRNA regulatory relationships. The package provides a complete set of pipelines for testing new methods and analysing new datasets. miRLAB includes a pipeline to obtain matched miRNA and mRNA expression datasets directly from TCGA, 12 benchmark computational methods for inferring miRNA-mRNA regulatory relationships, the functions for validating the predictions using experimentally validated miRNA target data and miRNA perturbation data, and the tools for comparing the results from different computational methods.
PLoS ONE
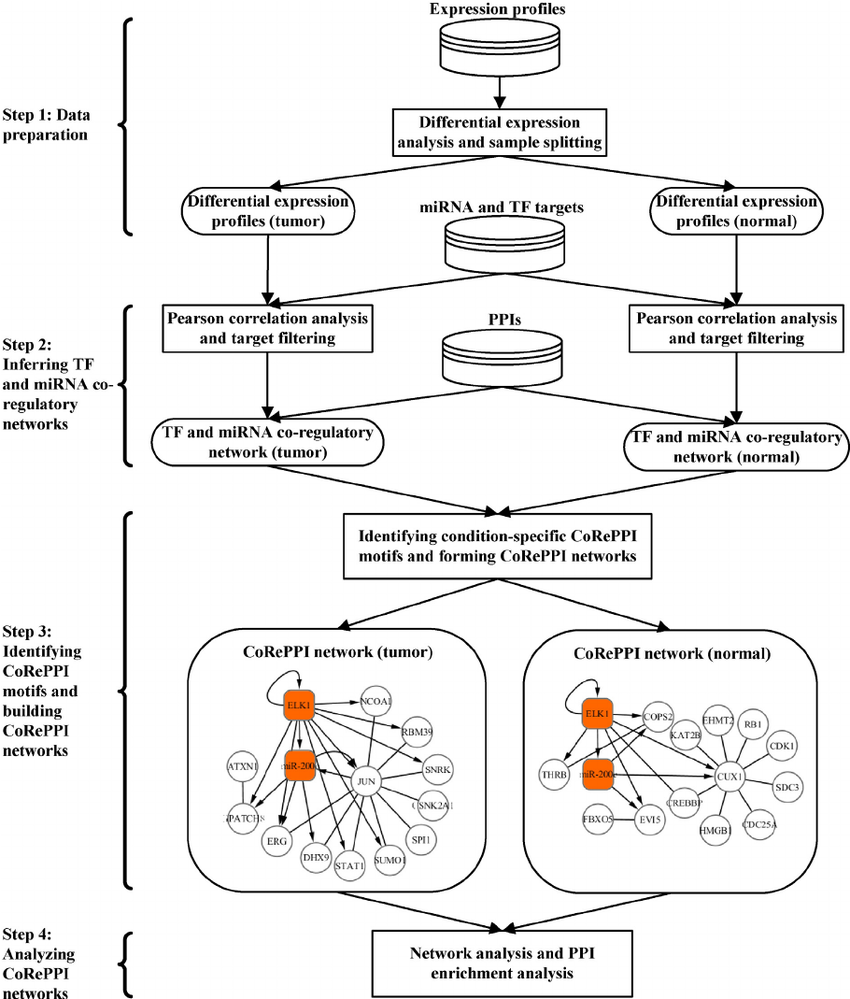
A novel framework for inferring condition-specific TF and miRNA co-regulation of protein-protein interactions
Recent studies have shown that transcription factors (TFs) and microRNAs (miRNAs), while independently regulate their downstream targets, collaborate with each other to regulate gene expression. However, their synergistic roles in protein-protein interactions (PPIs) remain mostly unknown. In this paper, we present a novel framework (called CoRePPI) for inferring TF and miRNA co-regulation of PPIs. Particularly, CoRePPI is aimed at discovering the co-regulation specific to a condition of interest, by using heterogeneous data, including miRNA and messenger RNA (mRNA) expression profiles, putative miRNA targets, TF targets and PPIs. CoRePPI firstly finds the network motifs indicating the co-regulation of PPIs by TFs and miRNAs in tumor and normal conditions separately. Then by identifying the differential motifs found in one condition but not in the other, it builds the networks consisting of TFs, miRNAs and their co-regulated PPIs specific to different conditions respectively. To validate CoRePPI, we apply it to the Pan-Cancer dataset which includes the expression profiles of 12 cancer types from TCGA. Through network topology analysis, we found that the tumor and normal CoRePPI networks are scale-free. Furthermore, the results of differential and intersected network analysis between the tumor and normal CoRePPI networks suggest that only a small fraction of the regulatory relationships between TFs and miRNAs are conserved in both conditions but they co-regulate different downstream PPIs in tumor and normal conditions; and in different conditions the majority of the regulatory relationships between TFs and miRNAs are different although they may regulate the same PPIs in their respective conditions. The CoRePPI sub-networks constructed for the three types of cancers (breast cancer, lung cancer and ovarian cancer) are all scale-free, and the intersection of these CoRePPI sub-networks can be utilized as the biomarker CoRePPI sub-network of the three types of cancers. The PPI enrichment analyses of the tumor and normal CoRePPI networks suggest that the co-regulating TFs and miRNAs are significantly associated with the specific biological processes, diseases and pathways. In addition, comparing with the two non-condition-specific approaches, the tumor CoRePPI network is found to have the most enriched cancer-related PPIs. Altogether, the results uncover the combined regulatory patterns of TFs and miRNAs on the PPIs, and may provide new insights for research in cancer-associated TFs and miRNAs.
Gene
Ensemble Methods for MiRNA Target Prediction from Expression Data
Background: microRNAs (miRNAs) are short regulatory RNAs that are involved in several diseases, including cancers. Identifying miRNA functions is very important in understanding disease mechanisms and determining the efficacy of drugs. An increasing number of computational methods have been developed to explore miRNA functions by inferring the miRNA-mRNA regulatory relationships from data. Each of the methods is developed based on some assumptions and constraints, for instance, assuming linear relationships between variables. For such reasons, computational methods are often subject to the problem of inconsistent performance across different datasets. On the other hand, ensemble methods integrate the results from individual methods and have been proved to outperform each of their individual component methods in theory.Results: In this paper, we investigate the performance of some ensemble methods over the commonly used miRNA target prediction methods. We apply eight different popular miRNA target prediction methods to three cancer datasets, and compare their performance with the ensemble methods which integrate the results from each combination of the individual methods. The validation results using experimentally confirmed databases show that the results of the ensemble methods complement those obtained by the individual methods and the ensemble methods perform better than the individual methods across different datasets. The ensemble method, Pearson+IDA+Lasso, which combines methods in different approaches, including a correlation method, a causal inference method, and a regression method, is the best performed ensemble method in this study. Further analysis of the results of this ensemble method shows that the ensemble method can obtain more targets which could not be found by any of the single methods, and the discovered targets are more statistically significant and functionally enriched. The source codes, datasets, miRNA target predictions by all methods, and the ground truth for validation are available in the Supplementary materials.
PLoS ONE
From miRNA regulation to miRNA-TF co-regulation: computational approaches and challenges
microRNAs (miRNAs) are important gene regulators. They control a wide range of biological processes and are involved in several types of cancers. Thus, exploring miRNA functions is important for diagnostics and therapeutics. To date, there are few feasible experimental techniques for discovering miRNA regulatory mechanisms. Alternatively, predictions of miRNA-mRNA regulatory relationships by computational methods have increasingly achieved promising results. Computational approaches are proving their ability as effective tools in reducing the number of biological experiments that must be conducted and to assist with the design of the experiments. In this review, we categorize and review different computational approaches to identify miRNA activities and functions, including the co-regulation of miRNAs and transcription factors. Our main focuses are on the recent approaches that use multiple data types for exploring miRNA functions. We discuss the remaining challenges in the evaluation and selection of models based on the results from a case study. Finally, we analyse the remaining challenges of each computational approach and suggest some future research directions.
Briefings in Bioinformatics
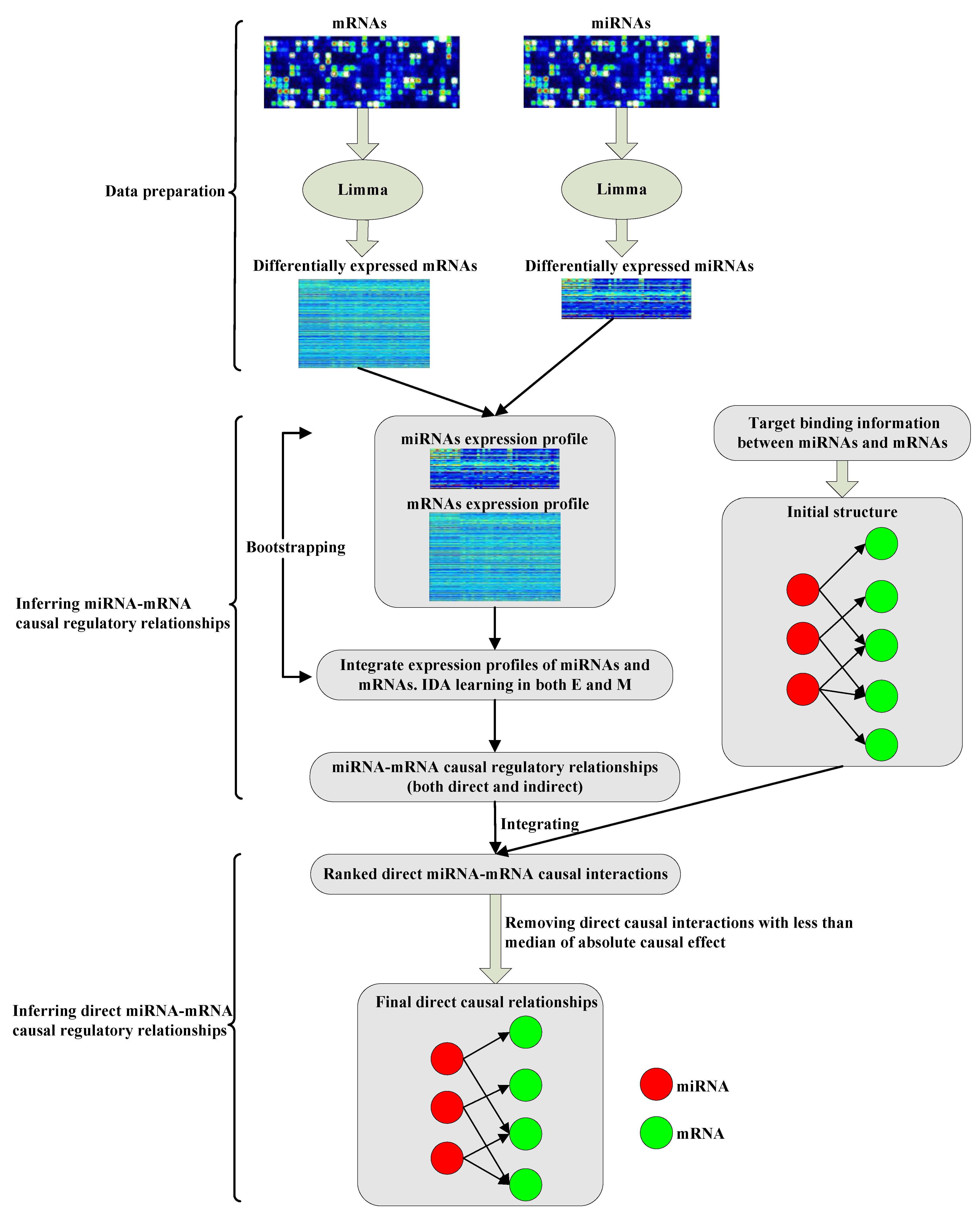
Identifying direct miRNA-mRNA causal regulatory relationships in heterogeneous data
Discovering the regulatory relationships between microRNAs (miRNAs) and mRNAs is an important problem that interests many biologists and medical researchers. A number of computational methods have been proposed to infer miRNA-mRNA regulatory relationships, and are mostly based on the statistical associations between miRNAs and mRNAs discovered in observational data. The miRNA-mRNA regulatory relationships identified by these methods can be both direct and indirect regulations. However, differentiating direct regulatory relationships from indirect ones is important for biologists in experimental designs. In this paper, we present a causal discovery based framework (called DirectTarget) to infer direct miRNA-mRNA causal regulatory relationships in heterogeneous data, including expression profiles of miRNAs and mRNAs, and miRNA target information. DirectTarget is applied to the Epithelial to Mesenchymal Transition (EMT) datasets. The validation by experimentally confirmed target databases suggests that the proposed method can effectively identify direct miRNA-mRNA regulatory relationships. To explore the upstream regulators of miRNA regulation, we further identify the causal feedforward patterns (CFFPs) of TF-miRNA-mRNA to provide insights into the miRNA regulation in EMT. DirectTarget has the potential to be applied to other datasets to elucidate the direct miRNA-mRNA causal regulatory relationships and to explore the regulatory patterns.
Journal of Biomedical Informatics
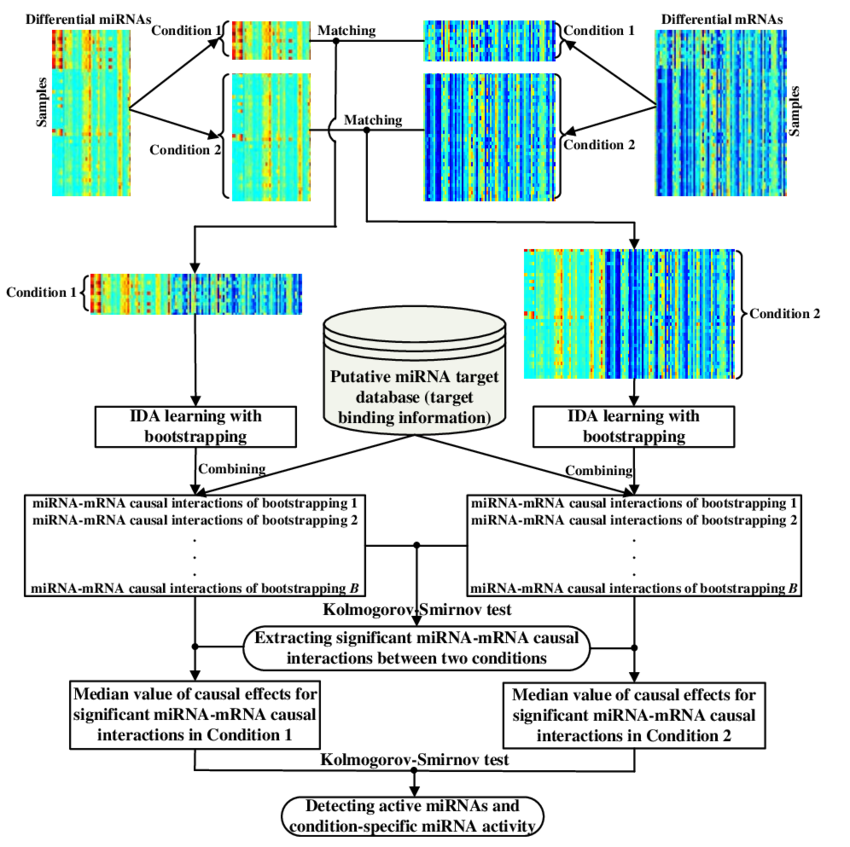
Inferring condition-specific miRNA activity from matched miRNA and mRNA expression data
MOTIVATION: MicroRNAs (miRNAs) play crucial roles in complex cellular networks by binding to the messenger RNAs (mRNAs) of protein coding genes. It has been found that miRNA regulation is often condition-specific. A number of computational approaches have been developed to identify miRNA activity specific to a condition of interest using gene expression data. However, most of the methods only use the data in a single condition, and thus, the activity discovered may not be unique to the condition of interest. Additionally, these methods are based on statistical associations between the gene expression levels of miRNAs and mRNAs, so they may not be able to reveal real gene regulatory relationships, which are causal relationships. RESULTS: We propose a novel method to infer condition-specific miRNA activity by considering (i) the difference between the regulatory behavior that an miRNA has in the condition of interest and its behavior in the other conditions; (ii) the causal semantics of miRNA-mRNA relationships. The method is applied to the epithelial-mesenchymal transition (EMT) and multi-class cancer (MCC) datasets. The validation by the results of transfection experiments shows that our approach is effective in discovering significant miRNA-mRNA interactions. Functional and pathway analysis and literature validation indicate that the identified active miRNAs are closely associated with the specific biological processes, diseases and pathways. More detailed analysis of the activity of the active miRNAs implies that some active miRNAs show different regulation types in different conditions, but some have the same regulation types and their activity only differs in different conditions in the strengths of regulation.
Bioinformatics
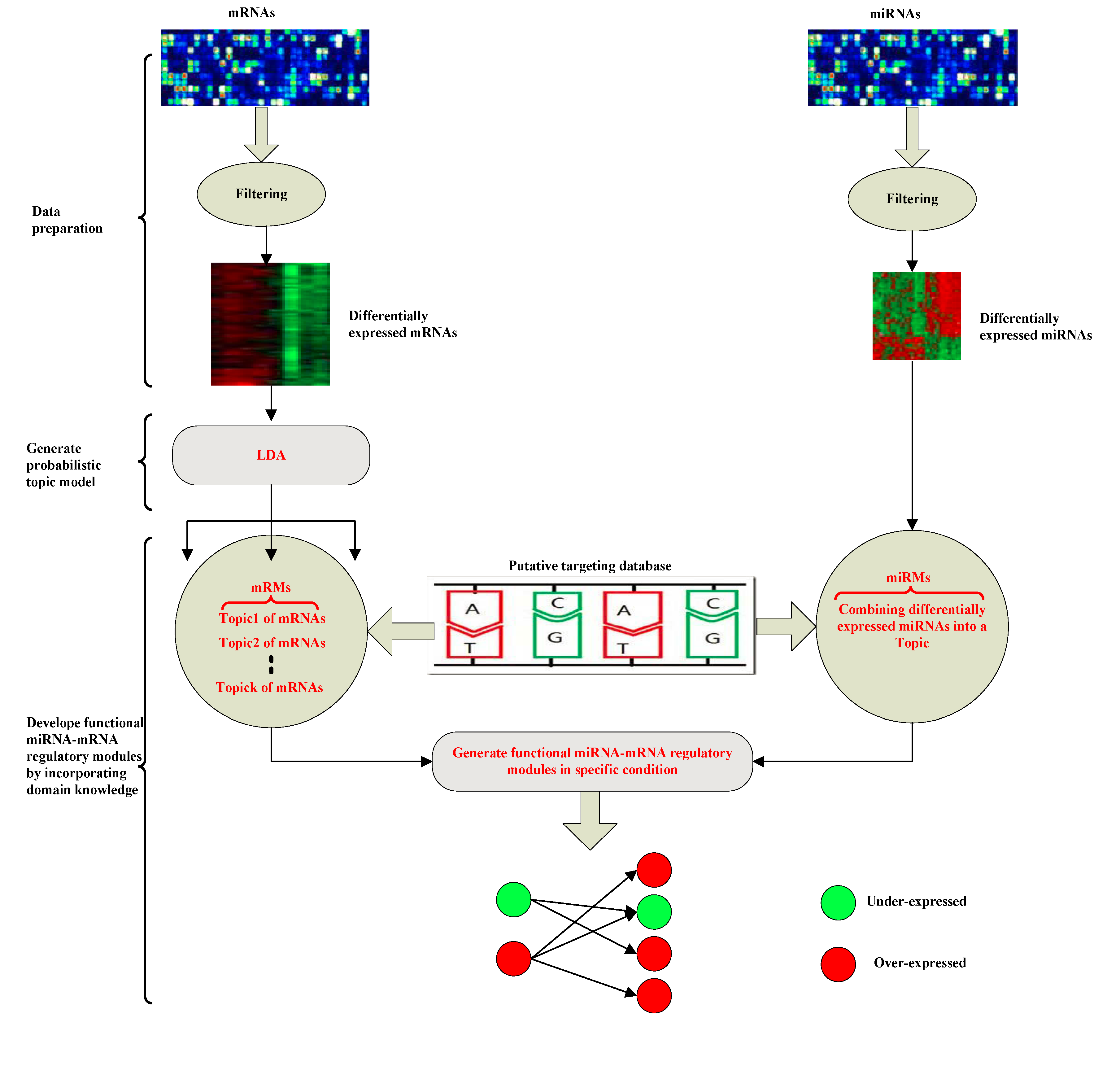
Inferring functional miRNA-mRNA regulatory modules in epithelial-mesenchymal transition with a probabilistic topic model
MicroRNAs (miRNAs) play important roles in gene regulatory networks. In this paper, we propose a probabilistic topic model to infer regulatory networks of miRNAs and their target mRNAs for specific biological conditions at the post-transcriptional level, so-called functional miRNA-mRNA regulatory modules (FMRMs). The probabilistic model used in this paper can effectively capture the relationship between miRNAs and mRNAs in specific cellular conditions. Furthermore, the proposed method identifies negatively and positively correlated miRNA-mRNA pairs which are associated with epithelial, mesenchymal, and other condition in EMT (epithelial-mesenchymal transition) data set, respectively. Results on EMT data sets show that the inferred FMRMs can potentially construct the biological chain of ‘miRNA→mRNA→condition’ at the post-transcriptional level.
Computers in Biology and Medicine